¿Qué es el síndrome de Marfan?
El síndrome de Marfan describe un complejo trastorno hereditario del tejido conectivo, que afecta principalmente a los ojos, el sistema cardiovascular y el sistema muscular esquelético. Sin embargo, considerando que cada órgano está formado por tejido conectivo, el síndrome de Marfan idealmente puede destruir e interferir fuertemente con el crecimiento y la función de cada sitio anatómico.
El síndrome se transmite como un rasgo autosómico dominante: por lo tanto, nos enfrentamos a una enfermedad genética grave, con una expresión fenotípica extremadamente variable (los defectos pueden diferir enormemente de una familia a otra o de un paciente a otro).
Lo que desencadena el síndrome de Marfan es la alteración del gen FBN1 (en el cromosoma 15), que codifica la fibrilina-1, una glicoproteína muy importante de la conexión que es el soporte estructural de las microfibrillas.
Microfibrillas: formadas por fibrilina, las microfibrillas están presentes en la matriz extracelular, en la que forman un entrelazamiento para la deposición de elastina en las fibras elásticas. Aunque son omnipresentes en el cuerpo, las microfibrillas abundan sobre todo en la aorta, los ligamentos y las zónulas de los cuerpos ciliares (nivel ocular).
Al ser un trastorno autosómico dominante, solo los niños que heredaron un gen FBN-1 alterado por ambos padres se ven afectados por el síndrome de Marfan. Sin embargo, en uno de cada 4 casos, la enfermedad es el resultado de mutaciones espontáneas en pacientes que no tienen antecedentes familiares.
El nombre de la enfermedad proviene del pediatra francés que la describió por primera vez en 1896 (A. Marfan), después de lo cual fue necesario esperar hasta 1991 para identificar el gen alterado involucrado en la manifestación sintomatológica: el descubridor fue F. Ramírez.
Mira el video
X Mira el video en youtubecausas
Hemos mencionado que el síndrome de Marfan es la expresión inmediata de la mutación de un gen que codifica para fibrilina-1. Intentamos profundizar el argumento para comprender qué mecanismo se establece para desencadenar el síndrome.
FIBRILLINE 1 es un componente glicoproteína de la elastina, esencial para garantizar y mantener la elasticidad y la resistencia del tejido. En condiciones fisiológicas, la fibrilina 1 se une a otra proteína, conocida como TGF-beta (o factor de crecimiento transformante beta). El TGF-beta parece estar involucrado en procesos nocivos del músculo liso vascular y la matriz extracelular. A partir de estos supuestos, algunos autores creen que el síndrome de Marfan se debe, además de la mutación del gen FBN-1, también a un exceso de TGF-beta, especialmente en la aorta, en las válvulas cardíacas y en los pulmones.
incidencia
Se estima que el síndrome de Marfan afecta a 1 sujeto cada 3, 000-5, 000 nacimientos y se manifiesta indiscriminadamente entre hombres y mujeres, sin predilección por una raza precisa. Las estadísticas muestran que el 75% de los pacientes tienen antecedentes familiares positivos; en el 25% restante, la causa reside en mutaciones esporádicas que parecen estar asociadas, de alguna manera, con la edad avanzada del padre en el momento de la concepción.
Los niños con formas extremadamente graves de síndrome de Marfan tienen una esperanza de vida de menos de un año.
Antes de la evolución de las estrategias de cirugía a corazón abierto, la mayoría de los pacientes con síndrome de Marfan tenían una esperanza de vida promedio de 32 años; Gracias a la mejora constante de las terapias médicas y farmacológicas, actualmente los pacientes con síndrome de Marfan viven en promedio hasta 60 años.
Signos y sintomas
Para profundizar: Síntomas del síndrome de Marfan
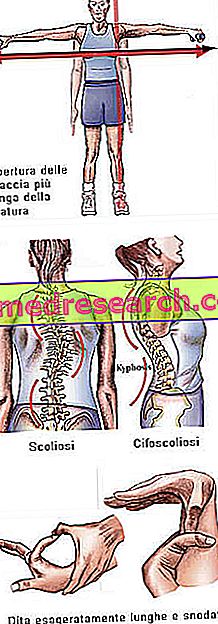
El síndrome de Marfan puede comenzar completamente asintomático. Los pacientes afectados tienen una estructura de línea exageradamente larga, siendo desproporcionadamente altos y delgados. Los miembros inferiores y superiores tienen una longitud mucho mayor que el tronco (dolicostenomegalia). También hablamos de aracnodactilia para expresar mejor el concepto de longitud exagerada de los dedos, típica de los afectados por el síndrome de Marfan: las manos se comparan con las patas de una araña.
Con respecto a la altura, estos pacientes son cortos, con un promedio superior al percentil 97.
Entre las otras características distintivas que se encuentran a menudo en pacientes con síndrome de Marfan, también mencionamos:
- Abertura de los brazos mayor que la altura.
- Articulaciones laterales → movilidad articular exagerada.
- Deformidad de la pared torácica.
- Dislocación de la lente.
- Parte superior del cuerpo menos desarrollada que la zona inferior.
- Neumotórax espontáneo (11%)
- escoliosis
- Estrías en la piel del muslo, espalda, deltoides, pectoral.
Entre los signos más problemáticos asociados con el síndrome de Marfan, recordamos el prolapso de la válvula cardíaca y la insuficiencia de la válvula mitral: una condición similar puede favorecer fácilmente la dilatación del anillo aórtico y la disección aórtica.
La tabla muestra los signos que se pueden encontrar en pacientes con síndrome de Marfan. Los caracteres descritos no siempre están presentes, pero se puede encontrar una buena parte de estos.
| Sitio anatómico afectado. | Posibles síntomas |
lindo | Estrías en las áreas torácica, lumbar y sacra. |
ojos | Alteración de la visión, astigmatismo, desprendimiento de la retina, glaucoma de ángulo cerrado, luxación del cristalino, miopía. |
Estructura ósea | Artralgia, cifoescoliosis, dolicostenomelia (desarrollo excesivo de la longitud de las extremidades en comparación con el tronco), hipermovilidad, paladar alto, pecho deformado, pies planos, muñecas estrechas y estrechas, reentrada / protuberancia anómala del esternón, escoliosis, hombros curvos, espondilolistesis |
Dita | aracnodactilia |
pulmones | Neumotórax espontáneo, disnea, enfermedad obstructiva pulmonar idiopática |
Alteraciones faciales | Paladar ogal (malformación del paladar), retrognacia mandibular (defecto del desarrollo de la mandíbula), cara alargada |
corazón | Angina de pecho, aneurisma aórtico abdominal, arritmia cardíaca, dilatación / rotura aórtica torácica, insuficiencia aórtica, prolapso de la válvula mitral |
idioma | Dificultad de lenguaje |
diagnóstico
Dadas las más de 200 posibles mutaciones, el uso de marcadores genéticos es casi imposible para fines de diagnóstico.
La evaluación del síndrome de Marfan no siempre es tan inmediata, ya que la expresión fenotípica de la mutación no siempre es evidente y fácil de identificar. El retraso en el diagnóstico puede comprometer seriamente la supervivencia del paciente: solo piense en la falta de reconocimiento de un problema cardiovascular.
Los criterios de diagnóstico para el síndrome de Marfan se redactaron internacionalmente en 1996: el diagnóstico consiste en una encuesta de antecedentes familiares asociados con una combinación de indicadores mayores y menores del síndrome.
Algunas de las muchas pruebas de diagnóstico utilizadas son:
- ecocardiograma
- Resonancia magnética angior y TC (para la investigación de la aorta)
- Angiografía por resonancia magnética (ARM) con líquido de contraste (para hacer visibles las estructuras aórticas internas)
- Examen con lámparas de hendidura (para analizar la posible dislocación de la lente).
- Medición de la presión ocular (para resaltar la posible presencia de glaucoma).
- Pruebas genéticas (recomendadas antes de concebir un niño para determinar o no el síndrome)
terapias
Al ser una enfermedad genética, no existe un medicamento o tratamiento que pueda revertir la enfermedad.
Sin embargo, el uso de medicamentos es esencial para aliviar los síntomas y evitar complicaciones, especialmente las cardíacas. Para este fin, los medicamentos para reducir la presión arterial, como los sartanes (especialmente), los inhibidores de la ECA y los bloqueadores beta, están particularmente indicados.
En el contexto del síndrome de Marfan, los pacientes que sufren de escoliosis también pueden recibir atención específica, así como a los afectados por el glaucoma.
La cirugía es concebible para corregir la dilatación aórtica anormal, un elemento que a menudo une a la mayoría de los pacientes con síndrome de Marfan.
Continuar: Síndrome de Marfan - Drogas y cuidados »


